
Our research group focuses on the study of the molecular mechanisms regulating cell death and inflammation, two interconnected cellular processes. Inflammation, the first response of the immune system to infection or tissue injury, is of crucial importance to protect the body against these insults. Nevertheless, inflammation needs tight regulation as it can turn into a maladaptive response at the origin of various diseases when not properly controlled. Cell death is essential during development and to maintain homeostasis in adults, but is also a major driver of inflammation, and consequenlty a suspected cause of human inflammatory pathologies. In the lab, we aim at getting a better understanding of the molecular mechanisms regulating the various froms of cellular demise, the exact nature of these suicide routes, and how dead cells drive inflammation. We study the molecular mechanisms that regulate the different cellular outcomes emerging from the innate immune Pattern Recognition Receptors (PRRs) and members of the Tumor Necrosis Factor Receptor (TNFR) family, namely cell survival, cell death and inflammation. We are interested in the role of autophagy in regulating these responses. Our studies are conducted at the biochemical, cellular and in vivo levels.
Areas of Expertise
- Cell Death
- Innate immunity
- Autophagy
- Signal transduction
- Post-translational modifications
Technology Transfer Potential
- Identification of new therapeutic targets for the treatment of cell death and inflammation related diseases
- Mouse models of autoinflammatory diseases
Selected publications
- van Loo, G. & Bertrand, M. J. M. Death by TNF: a road to inflammation. Nat Rev Immunol 23, 289-303 (2023). Visit ➚
- Huyghe, J. et al. ATG9A prevents TNF cytotoxicity by an unconventional lysosomal targeting pathway. Science 378, 1201-1207 (2022). Visit ➚
- Martens, A. et al. Two distinct ubiquitin-binding motifs in A20 mediate its anti-inflammatory and cell-protective activities. Nat Immunol 21, 381-387 (2020). Visit ➚
- Dondelinger, Y. et al. Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat Commun 10, 1729 (2019). Visit ➚
- Dondelinger, Y. et al. MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat Cell Biol 19, 1237-1247 (2017). Visit ➚
Bibliography
- Full bibliography Visit ➚
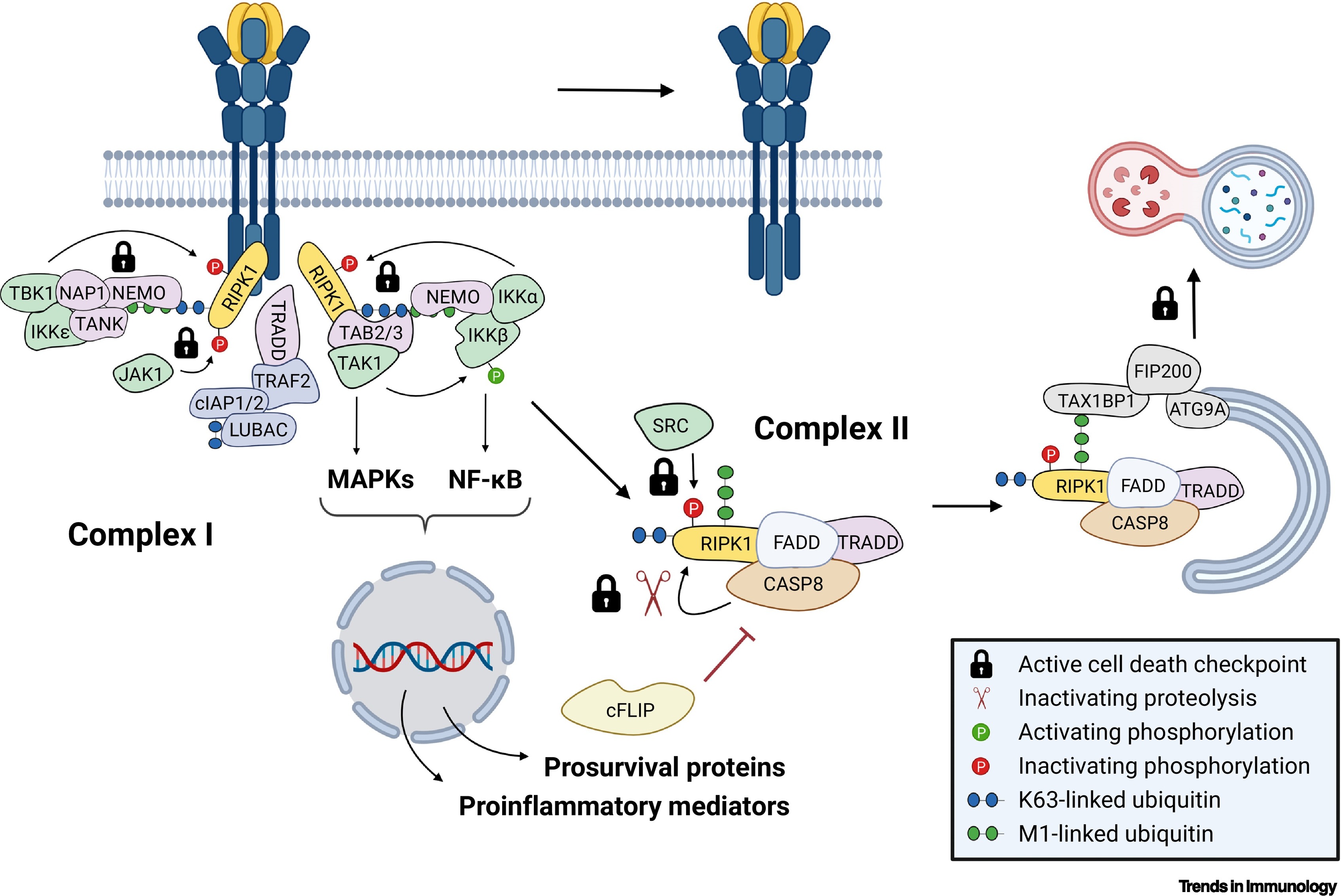
Normal cellular response TNFR1 activation by TNF. In most murine and human cell types, including macrophages and fibroblasts, sensing of TNF by TNFR1 does not trigger cell death but results in gene activation. TNFR1 complex I forms at the plasma membrane within seconds following TNF sensing. It serves as a recruitment and activation platform for the kinases that promote the activation of the nuclear factor κB (NF-κB) and mitogen-activated protein kinase (MAPK) signaling pathways resulting in the production of proinflammatory mediators and prosurvival proteins. Later, complex I destabilizes and detaches from the receptor to allow the formation of the caspase-8 (CASP8)-containing TNFR1 complex II in the cytosol. The cytotoxic potential of complex II is kept in check by several molecular mechanisms referred to as cell death checkpoints, which include its lysosomal degradation by an unconventional selective autophagy pathway. The cell death checkpoints that prevent the lethal activity of complex II are indicated in the figure as locks. For clarity the figure depicts only a subset of core constituents of the cell death checkpoints and the central components that are present in complex I/II. From Huyghe J, Priem D, Bertrand MJM. Trends Immunol. 2023